Education
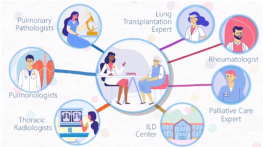
Interstitial Lung Disease
Free online continuing education on Interstitial Lung Disease! Get quality activities, articles, & videos to improve your Interstitial Lung Disease education.
Free online continuing education on Interstitial Lung Disease! Get quality activities, articles, & videos to improve your Interstitial Lung Disease education.


_263x0.png)