Education
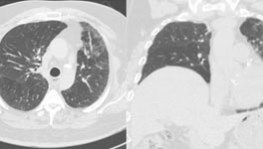
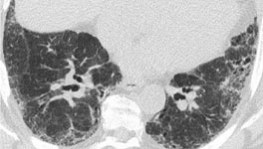
Idiopathic Pulmonary Fibrosis
Free online continuing education on Idiopathic Pulmonary Fibrosis! Get quality activities, articles, & videos to improve your Idiopathic Pulmonary Fibrosis education.
Results 1 - 58 of 58
Free online continuing education on Idiopathic Pulmonary Fibrosis! Get quality activities, articles, & videos to improve your Idiopathic Pulmonary Fibrosis education.